肠道和口腔微生物组在人与人之间传播的研究
The person-to-person transmission landscape of the gut and oral microbiomes

- 摘要 -
人类微生物组是人体的重要部分,也是许多健康状况的决定因素之一。然而,个体之间在多大程度上影响微生物组组成,以及微生物在人群中的传播仍然是未知的。在本篇文章里,利用9700多个人类宏基因组和菌株水平分析,我们检测到人与人之间存在广泛的细菌菌株传播,即具有明显的母亲到婴儿、家庭内和特定人口内的传播模式。在婴儿期,肠道微生物在母亲和婴儿之间传播是相当稳定的,并且在婴儿长大后仍可检测到。相比之下,口腔微生物组的传播主要发生在水平方向,并随着同居时间的延长而增强。同居个体之间会共享大量的菌株,肠道和口腔微生物组的菌种共享率分别为12%和32%,而且同居时间对菌株共享的影响大于年龄或遗传因素。共享的细菌菌株更好地再现了宿主群体结构。最后,特定的物种是有效的传播者,并与不同的预测细菌表型有关,而这些表型又与宿主外的生存能力有关。尤其是对于那些非感染性的、与微生物相关的疾病,本文所描述的微生物传播展示了其在人类微生物组研究中的相关性。
- 背景 -
我们的基因组遗传自父母,并且一生中保持稳定,只有有限的核苷酸变异。相比之下,我们的微生物(人类微生物组)构成也是从出生开始,但随着时间的推移而变化,显示出高度的时间变异性和个性化。众所周知,包括饮食和生活方式在内的许多因素都可以调节人类微生物组的组成,但由于很少微生物能在人体外生长,大多数微生物必须从其他个体获得。事实上,微生物在人类肠道的定殖主要是通过母体传播,但仅靠母体传播并不能解释成人微生物的多样性。微生物组是如何由个人获得并在人群中传播,以及如何形成个人微生物组仍然需要进一步的探索。到目前为止,由于研究数量和规模有限,仍然难以持续和全面地分析微生物的菌株,即物种内的遗传变异。
同一物种的菌株可以有高度的基因组和功能上的差异,基于菌株的分析是研究微生物传播的前提。确定微生物组传播将推进我们对人类微生物组复杂性的理解,并有助于研究微生物组传播对非传染性疾病的影响。因此,我们描述和量化了多种情况下人与人之间的微生物组菌株共享,从而全面描述微生物组的传播情况。
- 结果 -
微生物传播图谱
为了揭示人与人之间的微生物传播模式,我们使用菌株级别的宏基因组工具对一组大型的宏基因组数据集进行了综合分析,这些数据集具有已知的家族关系(n = 31)。其中8个数据集是在本研究中新测序的,它们来自有不同地理区域和宿主生活方式的美洲、非洲、亚洲和欧洲。其中非洲(加纳和坦桑尼亚)和欧洲(意大利)共收集了978份粪便和1,929份唾液样本。这个数据集共包括9715个微生物组样本(7646个粪便和2069个唾液)和经过整理的宿主信息,能够评估母婴、家庭成员、成年双胞胎、村庄和人口之间的传播。尽管这31个数据集的规模不同,且来自五大洲的20个不同国家,还代表了不同的宿主生活方式(图1a,b),但综合的数据集有利于在全球范围内研究人与人之间的微生物组传播模式。
本研究利用了一个经过验证的假设,从而通过宏基因组学进行微生物菌株传播推断。该假设是菌株通常在个人的肠道内至少持续几个月,但除非发生直接或间接传播,否则很难在没有关系的个体中发现。我们首先改进了菌种水平分析方法,然后进一步完善了菌种溯源,并进行了具体的物种定义。菌株分类是通过标准化的系统发育距离(nGD)阈值来定义的,该阈值能够将来自4个国家的1,500多个纵向样本中的同一个个体的纵向菌株保留,能够和不相关个体的nGD区分。在平均覆盖率相当低的情况下,这种基于nGD的阈值的方法在系统发育中表现良好。宏基因组样品中大多数可检测物种通常也具有这种特征(平均覆盖率=7.2×),并且连锁标记基因比对的长度有限(平均修剪比对长度=74,348核苷酸(nt))。此外,我们的方法还利用了系统发育树提供的进化模型信息。
通过对超过 214,000 个宏基因组组装基因组 (MAG) 和大约 138,000 个可用的单菌基因组分析,该微生物图谱扩展了 1,022 个尚未培养和未命名的物种(uSGBs),以及补充 1,730 个已经培养的物种 (kSGBs)。uSGBs在宏基因组样本中非常普遍,占所有检测到的物种级bins (SGBs) 的 37%。菌株共享是指样本SGBs中具有至少10%流行率的优势菌株,也至少在一个队列的至少20个样本中发现。结果共鉴定了来自肠道宏基因组中的646个SGBs,以及来自口腔宏基因组的252个SGBs,和两种环境中都存在的24个SGBs。开发的计算方法可用于推断菌株传播的任何宏基因组数据集。
以双歧杆菌 (SGB17256)为例,是646 个可传播的肠道 SGBs 之一,其在 1,298 个肠道微生物组样本(占粪便样本总量的 17%)中都进行了分析。有87%的来自同一个体的的成对样本中可以检测到相同的双歧杆菌菌株,这些样本的收集时间相隔六个月,菌株之间的 nGD 呈现明显的双峰分布(系统发育距离接近零的第一个峰表明共享菌株)(图1c)。总体而言,在绝大多数母亲及其后代,以及其他家庭成员之间,发现了 13,278 例个体共享双歧杆菌菌株。
尽管只有通过纵向取样或在特定环境(例如母亲到新生儿)下才有可能区分间接传播或直接传播,但我们通过在每个SGB中去除那些与MAGs或分离自发酵食品中的单菌具有高度相似性(≤0.0015 SNV 率)的菌株,降低从共同饮食来源中获得的共享菌株。尽管食物到肠道的定殖是罕见的,但由于食物微生物组的研究仍然很少,其他菌株或物种仍可能来自食物来源。这种过滤方法排除了肠道样品中可能来自于食品的大多数动物双歧杆菌 (SGB17278) 菌株。事实上,超过98%的排除样本来自西方化数据集,而在来自商业益生菌使用较少的非西方化数据集中仅检测到6个菌株。按照相同的标准,来自其他SGBs的540 个在系统发育上接近食物来源 MAG 的菌株被排除,包括嗜热链球菌、唾液链球菌和前庭链球菌 (SGB8002)(图1e)。总的来说,在排除这些菌株之后,我们在肠道样本中检测到不同个体之间约有 635 万个共享菌株,在口腔样本中检测到约 491 万个共享菌株。
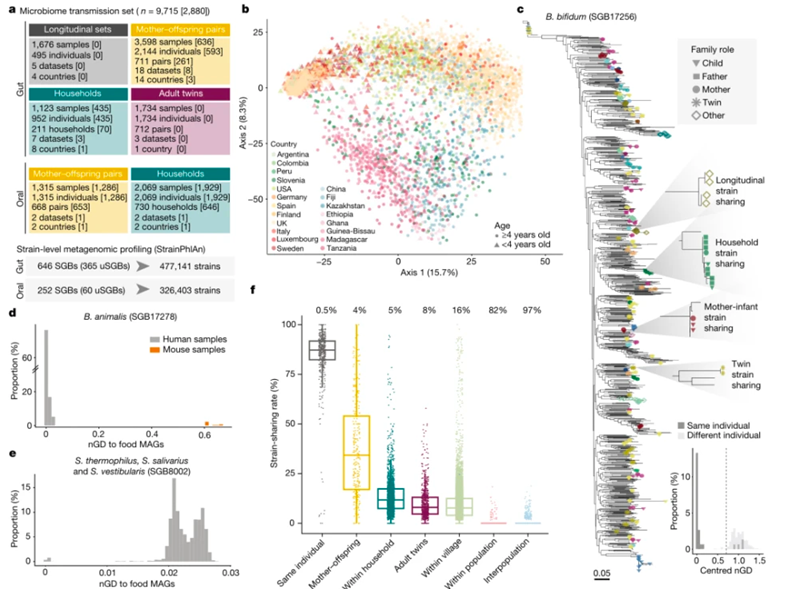
图1.研究人与人之间菌株传播的宏基因组框架。
(A) 概述基于SGBs的研究和数据集。(B)肠道样本的总体物种组成。(C) 双歧杆菌的系统发育树。(D) 动物杆菌(SGB17278)与从人类肠道宏基因组或小鼠样本中重建的菌株以及从发酵食品中重建的MAGs之间成对的nGDs分布。(E) 动物杆菌(SGB17278)与嗜热杆菌、唾液杆菌和前庭杆菌(SGB8002)之间的成对nGDs分布。(F) 不同关系的人与人之间的菌株共享率。
肠道微生物组传播概述
我们首先评估了人类关系中普遍的肠道微生物组菌株共享模式,将人与人之间的菌株共享率定义为通过SGBs数目标准化后的两个人之间共享的菌株数量。菌株在间隔不到六个月的受试者中具有高度持久性(平均 87% 的菌株共享率),只有 0.5% 的个体没有显示纵向重叠的菌株-可能是由于未报告的扰动或样本贴错标签的问题。一起居住的母亲及其0至3岁的后代之间检测到最高的菌株共享率(中位数为 34%),其次是同一家庭中4岁及以上的个体(12%)、非同居的成年双胞胎(8%)和非同居的同村成年人(8%)。成年双胞胎之间的共享菌株可能部分来自于母亲的传播,而同一村庄中个体之间的共享菌株可能是通过身体互动和微生物在环境中水平传播的结果。相比之下,不同村庄中的非同居人群显示出最小的菌株共享率(中位数 0%)。我们通过不共享菌株的个体百分比证实了这种显著的模式:只有 4% 的母子之间没有检测到菌株共享事件,而在同一没有明显接触的人群之间有82% 的比例没有共享菌株,另外不同种群中有高达 97% 的个体没有共享菌株(图1f)。因此,人与人之间的菌株共享基于社会距离,涉及共享的环境和亲属关系,这明显高于微生物差异所观察到的结果。总体而言,我们的综合分析强调了人与人之间的直接互动和社交互动网络在塑造单个个体肠道微生物组方面的相关性。
广泛的母婴传播
很多文章已经研究过母婴之间微生物组的传播,我们通过更大的样本集(来自711对母婴的3,598个样本,包括636个新的粪便样本;图1a)进一步报道了该模式。我们发现菌种共享率与子代年龄之间存在明显的负相关(Fig. 2a),尽管母子共享物种的数量随着后代年龄的增长而增加,表明后代可能有来自其他来源的物种。在生命的第一年,婴儿与母亲共享在婴儿和母亲微生物组中发现的一半菌株(菌株共享率),并且在婴儿中检测到的菌株有16%推定来自母亲,随着时间推移菌种共享率只有不明显的下降。这与断奶后的母子身体接触减少和婴儿运动活动范围扩大相一致。在1-3岁时菌株共享率下降到27%(图2a)。3岁以后,母子之间的菌株共享率趋于稳定(18岁以下为19%,30岁以下为14%;图2a),接近其他家庭成员之间的共享率(12%;图1f)。虽然出生时大量的菌株共享证实了母体微生物组存在于婴儿肠道,但在高龄个体(50-85岁)中,菌株共享仍然很重要,相对于没有关系的母亲,非同居的母子之间共享的菌株仍明显较多。这可能是出生时母体微生物和后来共同的社会环境的菌种传播共同作用导致的结果。
母体肠道微生物传播的潜在影响因素包括生活方式和分娩方式。尽管新测序的非西方人群的母亲和后代都有和西方化相关的微生物多样性的减少,我们注意到在大多数年龄组中母子菌株共享率没有很大差异。事实上,在西方和非西方的社区中,有类似数量的菌株通过母体传播。因此,非西方人群的微生物组的高度多样性似乎不是通过微生物母体传播来维持的,而可能是通过与更多个体的密切互动获得的。相比之下,我们确实证实了分娩方式与生命早期的母子菌株共享之间的关联:阴道分娩的婴儿(1岁以下)显示出与母亲的菌株共享率明显较高。然而,与年龄相关的分娩方式对婴儿微生物组的影响降低,3岁后没有发现差异。因此,虽然阴道分娩在生命早期通过母体传播影响了肠道微生物组,但生活方式的差异--包括卫生和建筑环境卫生水平的差异--并没有对微生物组传播率产生实质性影响。母亲到后代的传播(定义为1岁以下的后代--在菌种共享减少之前;图2a)在物种之间差异很大(图2b),但SGBs的传播率在各数据集之间相当一致,揭示了物种传播能力是微生物的一个特定特征。10个数据集中所有高度传播的SGBs都属于特征物种(kSGBs)(图2c),大部分属于拟杆菌属和双歧杆菌属(图2c)。比如,在所有西方的数据集中检测到普通拟杆菌(SGB1814)和长双歧杆菌(SGB17248)在母亲和婴儿之间传播(图2c)。相比之下,在婴儿中检测到的其他SGB--如在13名儿童和102名母亲中发现的Roseburiaintestinalis(SGB4951)则极少通过母体传播。在母亲和年长的后代之间发现高度母源性传播的SGBs逐渐减少(图2c),但即使在不与母亲同居的老年个体(50-85岁)中也检测到52%的高度母源性传播的SGBs(图2c)。

图2.母亲与子代的菌株共享。
(A)随着年龄增加,目前与子代的菌株共享率下降,而子代的物种丰富度增加。(B) 出生第一年的母婴SGB传播的分布。(C) 出生第一年母体传播较高的33种SGBs。
共同居住引起的传播
很多研究报道过家庭成员之间的肠道微生物组相似性,但由于缺少菌株级别的分析,大多数研究无法确定在更高的分类水平上是否能反映微生物的传播或评估一些条件的影响(比如遗传或饮食)。为了研究肠道微生物组的横向传播,我们评估了来自4大洲8个人群的212个家庭的883个同居者(4岁以下)的菌株共享情况(图1a),这些家庭的生活方式非常不同:从农村地区,到大型发展中国家和中等规模工业化富裕城市。大多数家庭显示出同居成员之间的菌株共享率(在11%和71%之间)明显高于同一人群中的非同居个体(图3a)。基于物种水平的微生物组相似性(β多样性指数),共同居住的家庭成员和非同居个人之间的差异较小。尽管人与人之间的菌株共享在很大程度上因家庭而异,但这只与西方化的生活方式略有关联,可能说明环境和社会的影响有限。同居者之间的菌株共享随着年龄的增长而减少。相比之下,非家庭来源的菌株(定义为不与任何家庭成员共享的菌株)的数量随着年龄的增长而增加。我们接下来评估了在四个已知亲属关系的人群中,父母和后代之间、兄弟姐妹之间以及伴侣之间的菌株分享。所有的家庭内部都显示出明显高于不同家庭之间的菌株共享率(图3b),但不同关系之间没有发现明显的差异。在4岁及以上的儿童中,母亲和父亲的菌种共享率相似,年轻有遗传关系的兄弟姐妹之间的菌种共享率略高于伙伴之间的菌种共享率(但不明显)。为了评估同居对生命后期菌种共享的影响,我们分析了过去曾住在一起的非同居成年双胞胎的宏基因组(1,734个样本,来自英国已发表的三个横断面数据集),包括单卵双生和双卵双生。考虑年龄影响后,双胞胎对之间的菌种共享随着分开生活的年数而明显减少(图3c)。除了过去同居的影响之外,还有一个遗传效应,单卵双胞胎在同居几十年后显示出比双卵双胞胎更高的菌株共享率。最后,不考虑双胞胎分开生活的年数,与年龄相关的共享菌种逐渐减少,这进一步证明了相对于遗传学和年龄,同居对成人微生物组传播的影响更大。因此,成年双胞胎之间的菌种共享可能更多的是过去同居的结果,而不是由于其父母传播微生物的长期影响。
来自10个不同细菌属的21种SGBs(占评估的SGB的4%)在家庭成员之间高度传播(图3d、e)。与母婴传播率相比,家庭内SGB传播性在不同的数据集中并不一致。我们观察到西方化和非西方化的生活方式之间的SGB传播率有很大差异(图3e),这与他们不同的微生物组组成一致。高度传播的SGBs中有很大一部分(38%)是没有特征分离株或基因组的物种(uSGBs)。家庭中广泛传播的双歧杆菌和拟杆菌属与那些在母亲和后代之间广泛传播的物种一致(图2c和3e),表明无论传播方式如何,这些都是有效的传播者,这与角双歧杆菌(SGB17231)相反,它出现了跨家庭传播。值得注意的是,在家庭内广泛传播的SGBs能够在分开居住的双胞胎之间共享(图3e),表明部分传播的菌株持续存在。

图3.家庭内部和家庭之间的肠道微生物传播。
(A)在至少有四个同居个体的72个家庭中的菌株共享率结果。(B) 家庭中个人之间的菌株共享率。(C) 非同居成年双胞胎的菌株共享率随着分开居住时间的增加而下降。(D) 家庭SGB传播率的直方图。(E) 在家庭中广泛传播的21种SGBs的传播率结果。
微生物在人群中的传播
与没有共同居住环境的个体相比,一个村庄中的非同居个体显示出肠道微生物组的菌种共享,尽管其比例明显低于同一家庭成员内部。尽管村内菌种共享率在人群中变化很大(图4a),但在67%的村庄中,来自同一村庄不同家庭的个体的菌种共享率明显高于不同村庄的个体。因此,人与人之间的微生物组传播也发生在远距离接触者之间的互动,并可能受到人口结构的影响。事实上,我们发现微生物组菌株在种群内和种群间的传播体现了宿主种群结构(图4b)。虽然只有4个SGB(0.8%)显示了较高的种群内传播率(图4c),但种群内内物种传播性在各数据集之间高度一致。几种广泛传播的SGBs是人类微生物组的已知成员:角芽孢杆菌(SGB17231,4%)、副血红链球菌(SGB8076,一种具有机会性病原体代表的物种、16%),以及嗜热链球菌等,表明与健康有关的和潜在的致病物种都可以成为有效的传播者。另外,瘤胃球菌科的一种未被鉴定的物种也属于高度传播的 SGB(SGB15073,1%)。尽管嗜热链球菌、唾液乳杆菌等在家庭中也广泛传播,但副血链球菌和SGB15073在非同居者中的特殊高传播性(图2c和3e)体现了不同的传播机制。

图4.基于村庄和人口的肠道微生物组的传播。
(A)一个村庄不同家庭的人与人之间的菌株共享率。(B) 显示人口结构的家庭数据集中的个人肠道微生物组共享菌株的无监督网络。(C) 不同家庭个体间高度传播的SGBs以及这些SGBs在特定数据集中的传播性。
横向的口腔传播
口腔微生物组菌株可能比肠道菌株更容易在个体之间传播,因为唾液可以成为直接载体,但目前人与人之间的口腔微生物传播仍然没有足够的研究。我们通过对252个SGBs进行菌种水平分析,评估了来自美国家庭的1,929个新测序的宏基因组(美国数据集)和来自斐济群岛的140个公开的唾液宏基因组中的口腔菌株共享模式。与观察到的肠道微生物组菌株共享率相似,我们研究了共享环境和具有亲属关系的人群的口腔菌种共享率:同居个体显示32%的口腔菌种共享率,而同一或不同种群的非同居个体分别共享3%和0%(图5a)。因此,同居个体的口腔菌株共享率比同一人群中的非同居个体高10倍,而物种水平的微生物组相似性则高不到0.5倍,这表明相比于相似的条件和生活方式,家庭成员之间的菌株传播更能影响微生物组组成。此外,不到0.5%的同一家庭成员不共享任何菌株,这表明人与人之间的口腔菌株传播比肠道微生物组传播更频繁(图1f)。另外结果显示了不同年龄和亲属关系的影响:与肠道微生物组模式相反,口腔菌株共享率随着后代年龄的增长而增加,特别是3岁以后,与后代口腔微生物组中微生物种类的累加相吻合(图5b)。此外在不同类型的关系中没有发现明显的差异,但伴侣之间的菌株共享率(中位数38%)略高于年轻后代(30%)和父亲(24%;图5a),这可能反映了更亲密的关系。在不同的年龄范围内,母亲与后代的物种共享率往往高于父亲与后代的物种共享率,这可能是通过母乳喂养密切接触的结果。然而,尽管与伴侣双方共享的菌株比例随着后代年龄的增长而略有增加(1岁以下为6%,18岁以下为8%;图5b),但与父母双方分别共享的菌株甚至更多(母亲为17-21%,父亲为13-17%)。总的来说,父母传播的口腔微生物菌株并不仅限在生命早期,似乎也取决于和接触时间有关的水平传播模式。
在很大程度上口腔菌株在家庭内部的传播因不同家庭而异(0-75%),虽然不能根据样本量不同的数据集得出和生活方式关联的结论,但我们确实发现在评估的所有类型的亲属关系中,家庭的菌株分享有明显的关联性(图5c)。因此,密切互动的家庭似乎有利于口腔微生物在所有同居成员之间的传播。我们接下来评估了父母与后代以及家庭内部的口腔微生物的传播性。来自16个不同属的18种SGBs(其中一半是uSGBs)在母亲和其1岁以下的婴儿之间显著高度共享(占评估的总SGBs的19%,图5d),包括两种普雷沃特菌和两种未表征的放线菌属(SGB17132和SGB17167)。尽管1岁以内的SGB传播性与1-3岁和3-18岁的SGB传播性表现出很强的相关性,只有5个物种在第一年龄段(1岁以下)和第二年龄段(1-3岁)之间持续广泛传播,3个物种持续到第三年龄段(3-18岁),后续还有多达68个物种出现(图5d)。这68个后来出现的物种与70个物种(包括28个uSGBs)高度一致,显示出明显的家庭传播性(占评估的SGBs总数的28%)。相比之下,没有任何物种在非同居个体中广泛传播。总的来说,以下三个口腔的SGBs表现出持续的传播能力:Actinomyces sp. ICM47 (SGB17167), Candidatus Saccharibacteria bacterium TM7 (SGB19822)和Flavobacteriaceae家族的uSGB (SGB2532)。

图5.口腔微生物组的传播。
(A)人与人之间的菌株共享率。(B)母子和父子之间的菌株共享率以及在子代中检测到的SGB数目。(C)不同的同居个体关系的菌株共享是正相关的。(D) 高度传播的 SGBs在不同年龄段的母亲和后代之间以及至少四岁的家庭成员之间持续存在。
与传播模式相关的表型
肠道微生物的传播性在具有一定地理距离且有不同生活方式的数据集中高度一致。同时,肠道微生物往往通过特定方式传播(图2c、3e和4c)。相比之下,不同传播模式下广泛传播的口腔SGBs基本上是重叠的(图5d)。物种的传播性似乎并不主要遵循一个传播模型--无论是相对丰度还是种群中的流行率都与其传播能力呈正相关。普遍性和可传播性之间没有直接联系,这与物种通过不同方式传播相一致,因此我们接下来探讨了与环境持久性相关的表型特性是否能更好地解释我们检测到的模式。由于我们菌株水平上分析的58%的肠道和24%的口腔SGBs尚未培养,我们根据它们的基因组序列推断细菌的表型。预测的表型与实验确定的性状有90%以上一致性。肠道和口腔微生物组的传播模式与特定的表型有关(图6)。革兰氏阴性菌--通常对清洁剂和消毒剂更具抵抗力,显示出更强的肠道和家庭传播能力,同时家庭内口腔微生物的传播性也增加。远距离的肠道微生物传播需要更强大的环境生存机制--即空气性和孢子形成。不到10%的肠道SGBs被预测为耐氧,而超过66%的口腔SGBs被预测为耐氧。最后,从母亲传给后代的频率来看,婴儿肠道中运动性的物种低于非运动性SGBs,鉴于运动性和毒力之间的联系,这可能是有益的。总的来说,我们的结果表明,与环境中生存相关的微生物表型特征部分影响了人与人之间的肠道微生物传播,而与口腔微生物传播的联系明显较弱。

图6.肠道和口腔微生物传播性和表型特征之间的关联。
- 讨论 -
通过对跨不同人群的微生物传播的综合多队列研究表明,人与人之间存在广泛的微生物传播。这证实了已经提出的假设,并揭示了菌株在长期密切接触的个体之间的转移影响了个人微生物组成,从而影响了可能的代谢和宿主-微生物相互作用。正如预期的那样,在生命的第一年,母亲和婴儿的肠道微生物组之间的菌株共享是最大的,共享的菌株也分别占同居个体之间肠道和口腔微生物组的12%和32%(图1f和5a),这种影响可能是由密切的身体互动引起的,即使这种互动在成年后才开始(伴侣之间的肠道和口腔菌株共享率分别为13%和38%;图3b和5a),并且在很长一段时间内是部分可逆的,双胞胎在分开生活30年后,其菌株共享率从最初的30%减少到约10%(图3c)。由于不同人群甚至同一人群中不同村庄的无亲属关系的个体几乎不共享任何菌株(菌株共享率中位数为0%),我们的结果强调了社会互动对微生物组有不可忽视的影响,这可能在微生物组相关疾病中发挥作用,意味着需要在人类微生物组研究中考虑人与人之间的菌株传播。相比之下,我们发现不同的生活方式对微生物组传播的影响很小:尽管根据饮食、医疗设施和药物的使用以及卫生条件等特征,将存在巨大的微生物组组成差异的人群定义为西方化和非西方化的人群,但我们发现垂直和水平菌株共享率非常相似。要确保这一结果的可靠,需要更大的、多样化的队列和关于参与者生活方式和文化习俗的更详细的基础数据,但我们的结果可能表明不同人群中类似的微生物定殖抗性,这可能对建立持久的定殖更为重要。我们的结果还表明,在非西方化社区观察到的较高的微生物丰富度并不是由其他家庭成员的传播导致的,而是与环境以及支持微生物多样性的饮食和生活方式相互作用的结果。显示出特别高的传播性的物种(图2c、3e、4c和5d)应该成为深入了解基因组和表型特征的起点,这些特征反过来可以为传播机制提供信息。尽管我们的研究不能解决人与人之间的微生物组传播是否是直接传播及其方向性,但它提供了一个关于人类微生物组传播的系统概述。设计特定的研究来模拟人类常规社交网络的变化(例如,在家庭变化之后)或其他社会动物,可以获得对人与人之间微生物组传播及其方向性的进一步了解。我们所使用的改进的菌株追踪方法,包括迄今未培养物种的菌株分析和基于系统发育距离的特定菌株的定义,使我们扩展到超过80多万个菌株的样本。尽管如此,未来通过更深入的测序、长读长技术或单细胞方法实现全基因组分辨率的研究可能会进一步完善这些发现。总的来说,我们的结果强化了这样一个假设:考虑到传播性和社会网络结构将改变未来微生物组研究,应该重新评估目前被认为是非传染性的几种疾病。
参考文献
Valles-Colomer, M., Blanco-Míguez, A., Manghi, P. et al. The person-to-person transmission landscape of the gut and oral microbiomes.Nature(2023).










{kind=link}
在线咨询